
Research Interests
Chromatin, the mix of DNA and proteins that forms chromosomes within the nucleus of humans and other higher organisms, plays a critical role in gene regulation and in turn a central role in development and disease. Indeed, pathogenic mutations in the genes that encode enzymes that methylate DNA or histones, the building blocks of chromatin, have been identified in numerous developmental disorders as well as cancer (see Nature Reviews Genetics, Janssen & Lorincz, 2022). Research in the lab is directed towards understanding the molecular interplay between transcription, DNA methylation and post-translational modifications of histones. Towards this end, we use both human and mouse tissue culture models as well as genetically engineered mice. Ultimately, our goal is to characterize the mechanistic basis of “epigenetic disorders”, including neurodevelopmental chromatinopathies, such as Sotos, Weaver and Rahman Syndromes and cancers in which the genes that encode chromatin factors are mutated. We employ CRISPR/Cas9, degron technology, conditional knockouts using Cre/loxP, and conventional genetic knockouts of clinically relevant chromatin factors or regulatory regions alongside genome-wide analyses of gene expression and chromatin features to dissect the roles of specific epigenetic marks and their  interplay in gene regulation. Genome-wide tools employed include low-cell input methods for whole genome analysis, such as for transcription (RNAseq), DNA methylation (PBAT) and chromatin marks (ULI-ChIP-seq developed by Julie Brind’Amour, former postdoc in the lab). Analyses of these datasets are carried out using published and in house pipelines developed to integrate the analyses of these epigenomic datasets, including at an allele-specific level (MEA, developed by Julien Richard Albert, former graduate student in the lab).
interplay in gene regulation. Genome-wide tools employed include low-cell input methods for whole genome analysis, such as for transcription (RNAseq), DNA methylation (PBAT) and chromatin marks (ULI-ChIP-seq developed by Julie Brind’Amour, former postdoc in the lab). Analyses of these datasets are carried out using published and in house pipelines developed to integrate the analyses of these epigenomic datasets, including at an allele-specific level (MEA, developed by Julien Richard Albert, former graduate student in the lab).
Ongoing projects
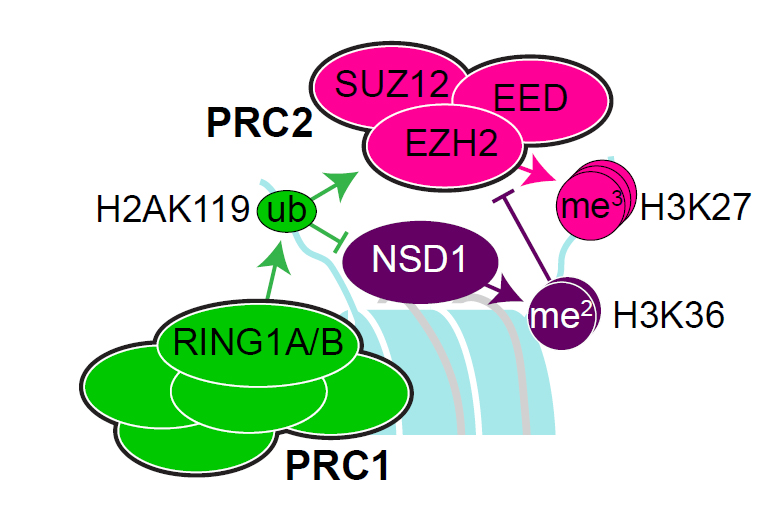 1) Sotos and Weaver Syndromes are related prenatal overgrowth disorders resulting from pathogenic loss-of-function mutations in the chromatin factors NSD1 and EZH2, which deposit covalent histone modifications on the core histone H3, specifically at lysine 36 (H3K36me2) and lysine 27 (H3K27me3), respectively. Using mouse embryonic stem cell models of these deficiencies, our goal is to dissect the interplay between these epigenetic marks in early embryonic development and the impact of their disruption on gene expression. Defining the role of these epigenetic factors during this critical period of development will hopefully yield new therapeutic approaches for treatment of these disorders.
1) Sotos and Weaver Syndromes are related prenatal overgrowth disorders resulting from pathogenic loss-of-function mutations in the chromatin factors NSD1 and EZH2, which deposit covalent histone modifications on the core histone H3, specifically at lysine 36 (H3K36me2) and lysine 27 (H3K27me3), respectively. Using mouse embryonic stem cell models of these deficiencies, our goal is to dissect the interplay between these epigenetic marks in early embryonic development and the impact of their disruption on gene expression. Defining the role of these epigenetic factors during this critical period of development will hopefully yield new therapeutic approaches for treatment of these disorders.
2) Rahman/hist1h1e syndrome, which shares characteristics of Sotos and Weaver Syndromes, results from pathogenic frame shift mutations in the gene encoding the linker histone H1.4 that yield a common novel C-terminal region. We are currently using an unbiased proteomic approach to catalog the chromatin factors that interact with wildtype and mutant H1.4 and will characterize the impact of these H1.4 mutations on the epigenome and as well as in gene regulation, with the goal of defining the molecular basis of this overgrowth syndrome.
3) DNA methylation plays a critical role in development and disease, with congenital mutations in each of the DNA methyltransferases yielding distinct disorders. Recent studies suggest that the covalent histone modification H3K9 trimethylation (H3K9me3) plays an important role in directing the maintenance DNA methylation machinery to specific genomic sites. Specifically, the nuclear factor UHRF1, which is required for DNA methylation maintenance following DNA replication, includes a domain that recognizes/binds to H3K9me3. Our aim is to determine whether such binding is required to promote DNA methylation of transposable elements and imprinted regions during early embryonic development, when DNA methylation levels are generally low across the rest of the genome.